What to Know About Amyloidosis
Symptoms, Causes, Diagnosis, and Treatment
HengDao / Getty Images
Medically reviewed by Jay N. Yepuri, MD, MS
Amyloidosis is a rare disease in which amyloid protein builds up in the organs. Amyloid is an abnormal version of a protein normally produced by the body. It is not easily broken down.
Amyloidosis can affect various organs, including the heart, kidneys, liver, and spleen, the nervous system, and the digestive tract. The buildup of this protein in the organs will cause them to not work correctly.
Amyloidosis has no cure and, without treatment, can lead to organ failure. Fortunately, the condition is treatable, and its symptoms and amyloid production can be managed.
This article explains what amyloidosis is, including symptoms, causes, types, and more.
HengDao / Getty Images
What Is Amyloidosis?
Amyloidosis is sometimes called a protein misfolding disorder, in which misfolded proteins are the main reason the condition develops. The specific protein involved differs between types of amyloidosis.
The normal versions of amyloid proteins typically have jobs in the body, from supporting immunity to regulating fluid balance and body processes. When the normal proteins finish their assigned jobs, they leave the bloodstream. But with amyloidosis, they misfold, take on abnormal shapes, and get deposited into the organs.
Amyloidosis is either systemic (widespread or affecting the whole body) or localized to one body area, and there are multiple subtypes.
Systemic amyloidosis is more common and can affect multiple organs and body tissues. Left untreated, it can lead to organ damage. Localized amyloidosis will affect one organ or only one area of the body.
Amyloidosis Symptoms
The symptoms of amyloidosis start subtly and will vary depending on where amyloid proteins collect.
Symptoms might include:
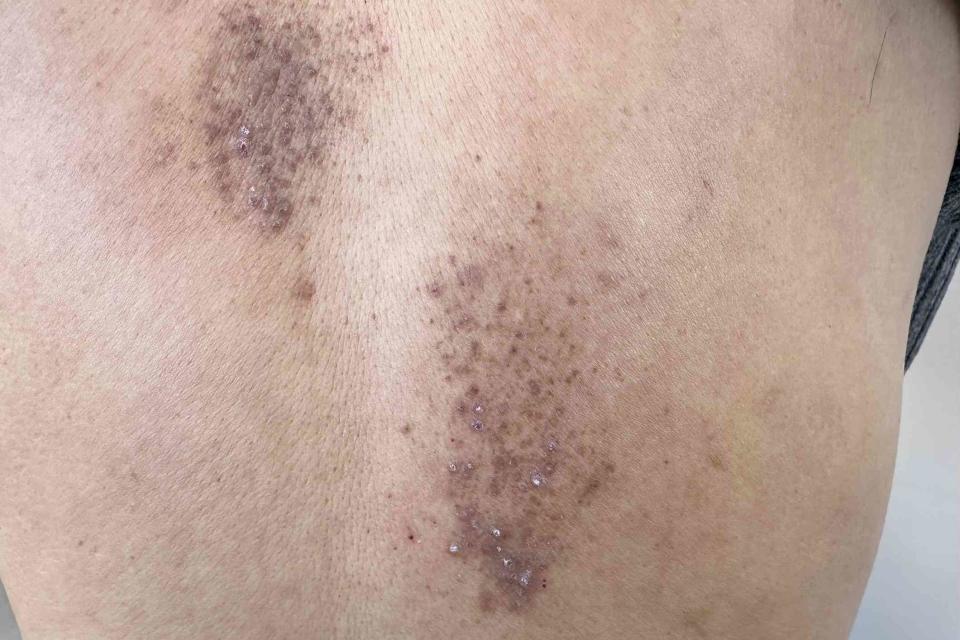
Skin changes, including a waxy thickening, bruising on the chest, face, and eyelids, and purple patches around the eyes
Severe fatigue
Dizziness or near fainting when standing because the nerves that control blood pressure are affected
Numbness, tingling, weakness, or pain in the hands or feet—this is because amyloid proteins collect in the nerves of the fingers, toes, and soles of the feet
Urine changes (including foamy urine) when amyloidosis damages the kidneys, causing proteins to leak from the blood into the urine
Swelling in the legs, feet, ankles, and calves
Diarrhea or constipation if amyloidosis affects the nerves that control the bowels
Digestive system troubles that affect digestion and absorption of nutrients
An enlarged tongue if the condition affects the tongue muscles
Muscle enlargement, especially around the shoulders
Unintentional, significant weight loss due to protein loss and loss of appetite
Irregular heartbeat, shortness of breath even with light activity, and/or signs of heart failure if amyloidosis affects the heart
Related: What Is Cardiac Amyloidosis?
Because some types of amyloidosis can affect more than one organ or multiple tissues, you may experience symptoms that affect more than one body area. It is also possible to not have any symptoms at all, especially early on.
What Causes Amyloidosis?
Amyloidosis occurs when amyloid protein builds up in one or more of your organs and causes them not to work correctly. It is sometimes secondary to another health condition or can develop as a primary condition.
Amyloidosis can also occur in people on long-term dialysis. Sometimes, it is due to a gene mutation, but other times, the cause is unknown.
Gene abnormalities linked to amyloidosis are present at birth and associated with an increased risk for the condition. Additionally, the type of gene abnormality can lead to disease complications or affect the age at which symptoms appear and how the condition progresses.
No Way to Prevent Amyloidosis
Aside from genetics, scientists do not actually know what causes amyloidosis or why certain proteins become misfolded. That means there is nothing a person can do to prevent amyloidosis. And while researchers are able to point to specific genes that might cause the disease, genes cannot be changed.
Types of Amyloidosis
The amyloidosis type is identified by the specific proteins that form amyloid deposits and where amyloid protein accumulates. Some types are life-threatening, while others cause less harm.
The most common types of amyloidosis are light-chain (AL) amyloidosis, autoimmune (AA) amyloidosis, beta-2 microglobulin amyloidosis (Abeta2m), Transthyretin (ATTR) amyloidosis, localized amyloidosis (ALoc), and wild-type ATTR.
Light-Chain Amyloidosis
Light-chain amyloidosis—also called primary amyloidosis—is the most common type, accounting for 70% of people with amyloidosis. The body’s immune system produces abnormal antibodies called light chains with AL amyloidosis.
Normal plasma cells (a type of bone marrow cell) produce antibodies (proteins that fight infections). But sometimes, plasma cells can produce extra pieces of antibodies called light chains that misfold and bind together to form amyloid fibers. Amyloid proteins will circulate in the bloodstream and deposit throughout the body.
Autoimmune (AA) Amyloidosis
Autoimmune amyloidosis (AA)—also called secondary amyloidosis—occurs due to a reaction from a chronic inflammatory disease or a chronic infection. This type of amyloidosis affects the kidneys about 80% of the time.
The protein involved in this type is serum amyloid A, which is normally a part of the inflammatory reaction. Generally, an inflammatory reaction breaks down amyloid proteins, but this does not always happen in people with chronic inflammatory diseases. People with rheumatoid arthritis (RA) and inflammatory bowel disease (IBD) have the highest risk for secondary amyloidosis.
AA amyloidosis is also linked to hereditary conditions that disrupt gene processes, such as familial Mediterranean fever, which causes fever episodes accompanied by abdomen, chest, or joint pain.
Beta-2 Microglobulin Amyloidosis (Abeta2m)
Abeta2m affects people with chronic renal failure who have been on dialysis for many years. Beta-2 microglobulin is an immune system protein normally found on the surface of cells and in small amounts in the blood and urine. Here, amyloid deposits of beta-2 microglobulin accumulate in the joints and tendons because the kidneys cannot expel them.
Transthyretin (ATTR) Amyloidosis
ATTR is a type of inherited amyloidosis. It runs in families and is caused by a mutation of the transthyretin (TTR) gene that leads to abnormal transthyretin protein. This protein normally transports thyroid hormone (thyroxine) and retinol (vitamin A) to the liver.
The abnormal TTR proteins are amyloid fibers that, in excess, can lead to neuropathy and cardiomyopathy in middle age or late in life.
Localized Amyloidosis (ALoc)
There are different types of localized amyloidosis. ALoc leads to localized amyloid deposits in the airway, skin, eyes, and urinary tract. Amyloid deposits result from a localized production of immunoglobulin light chains and not ones that originate in the bone marrow plasma cells.
Wild-Type ATTR
Wild-type ATTR is related to aging, and often affects males over age 75. The amyloidosis type affects the heart and causes carpal tunnel syndrome as an early symptom.
Amyloidosis Diagnosis
Early diagnosis of amyloidosis can prevent organ damage, but the condition is often overlooked because symptoms can mimic those of other conditions. Therefore, it is helpful to share as much information as possible with your healthcare provider to assist them in making a diagnosis.
Your healthcare provider will start with a physical exam. They will also ask about your medical history, including symptoms, to determine what additional tests might be needed.
Laboratory tests and imaging can help make an amyloidosis diagnosis. These may include:
Blood and urine testing: Both blood work and urine testing can check amyloid protein levels. Blood tests can also check thyroid and liver function.
Biopsy: With a biopsy, your healthcare provider will remove a sample of tissue from the liver, kidneys, nerves, heart, or another organ to figure out what type of amyloid deposits you may have.
Bone marrow biopsy: A bone marrow biopsy removes tissue from inside the bone. These samples are then sent to a lab to check for abnormal cells.
Echocardiogram: This imaging test uses sound waves to take pictures of the heart.
Magnetic resonance imaging (MRI): MRI imaging can reveal detailed images of the organs and body tissues. MRI can also be used to view the structure and function of your heart.
Nuclear imaging: This imaging type uses radioactive material injected in a vein to view the heart and look for damage or signs of amyloidosis. It can also help your healthcare provider determine the type of amyloidosis.
Immunohistochemistry (IHC) can be used to subtype amyloidosis. This test involves tissue collection to look for specific amyloid proteins on the surface of cells. A protein electrophoresis test is sometimes done in combination with the IHC test. The protein electrophoresis test can detect most amyloidosis conditions.
Amyloidosis Treatment
Amyloidosis is a chronic condition without a cure, but treatment can manage symptoms and limit amyloid production. Treatment can vary based on the type of amyloidosis and symptoms.
With secondary amyloidosis or autoimmune type, treatment starts with managing the underlying disease. For example, if the underlying cause is RA or IBD, the condition is treated with biologics and other disease-modifying antirheumatic drugs (DMARDs). In this case, symptoms ease when inflammation goes down.
Your healthcare provider may also prescribe medicines that help to clear amyloid fibers from the bloodstream, including corticosteroids, chemotherapy, and immunotherapy drugs.
For hereditary amyloidosis types, liver transplantation is the most effective way to slow down the disease. A new liver will not produce abnormal amyloid proteins. Researchers are also looking at new therapies to prevent amyloid proteins from being deposited in the organs of people who develop this hereditary subtype.
Treatments for primary amyloidosis include corticosteroids and chemotherapy medicines, such as Velcade (bortezomib). These therapies can slow down organ damage, improve quality of life, and even prolong life.
Researchers are investigating high-dose chemotherapy with autologous stem cell transplantation for treating primary amyloidosis and prolonging survival. Autologous stem cell transplantation involves harvesting a person’s own stem cells from their blood. Once they have high-dose chemotherapy, the stem cells are returned to populate the bone marrow.
A person whose kidneys have been damaged by amyloidosis may need dialysis to filter wastes, salts, and other fluids from the blood regularly. Other subtypes of amyloidosis are treated by managing symptoms and with medicines that help to clear amyloid fibers from the bloodstream.
Complications Associated With Amyloidosis
Treatment for amyloidosis can decrease symptoms and prevent disease-related complications. Complications associated with amyloidosis include kidney and heart failure and nervous system problems, as follows:
Kidney failure: Amyloid proteins can affect the kidneys’ ability to remove waste products from the body, eventually leading to kidney failure.
Heart failure: Amyloid proteins reduce the heart’s ability to correctly fill with blood as the heart beats, which leads to less blood with each heartbeat. It can also affect the heart’s electrical system leading to heart rhythm problems. Amyloidosis heart problems can be life-threatening if not addressed.
Nervous system problems: Amyloidosis can cause nerve damage, pain, numbness, and tingling in the hands and feet. It might also affect the nerves that control blood pressure and bowel function.
Who’s at Risk for Amyloidosis?
Certain risk factors can predispose a person to amyloidosis. These may include:
Age: Most people diagnosed with amyloidosis are between ages 50 and 65, although people as young as 20 can develop this condition.
Other diseases: People with chronic infections and inflammatory diseases have an increased risk for amyloidosis.
Family history: Some amyloidosis types are hereditary.
Race: People of African descent have an increased for carrying a genetic mutation associated with some amyloidosis types.
Kidney dialysis: In people on dialysis, abnormal amyloid proteins can build up in the blood and become deposited into tissue.
Multiple myeloma: Around 10% to 15% of people with multiple myeloma will develop AL amyloidosis. Multiple myeloma is a type of cancer that forms in the plasma cells.
Related: M-Protein Antibodies and Significance in Blood
Outlook and Life Expectancy
The most significant predictor for outlook and life expectancy with amyloidosis is how much of the heart is affected by amyloid proteins. Damage to the heart, kidneys, gastrointestinal tract, and nervous system can also affect both outlook and life expectancy.
Life expectancy can vary based on the type of amyloidosis and how successful the treatment might be. Some types are life-threatening or fatal, while others are easily treatable.
For example, AL amyloidosis treatment can help people to live longer and even lead to long-term remission (periods without disease symptoms). In one study of 46 people with AL amyloidosis, 33% of people who took dexamethasone and melphalan achieved complete remission. The disease would be fatal without appropriate or successful treatment.
Successful treatment of the underlying cause can lead to a good outcome for people with AA amyloidosis. The earlier a person is diagnosed, the better their outlook will be. Early treatment can also reverse amyloid deposits and prevent kidney damage.
The specific genetic mutations linked to ATTR amyloidosis improve or worsen disease outcomes and life expectancy. The most common gene mutation for the disease is V30M, and people with this mutation respond well to liver transplants, improving life expectancy. Additionally, if the disease is left untreated, it could be fatal after around 7 to 10 years.
People with wild-type ATTR generally have better outcomes than they would with AL amyloidosis and ATTR amyloidosis. According to one 2015 study, 85% of people with wild-type survived past their one-year follow-up. People who experienced severe heart problems were more likely to have worse outcomes, but having a heart transplant significantly increased life expectancy.
It is important to understand that many factors determine life expectancy with amyloidosis, and much of the information available is related to averages and are not specific to personal outcomes. Your outlook is likely to be different than someone else's.
And research has produced significant advancements in early diagnosis and successful treatment of amyloidosis, so a long and good-quality life is possible even with the condition.
Questions About Outlook and Survival
Contact your healthcare provider with your concerns or worries about your survival or outcomes. They are best positioned to help you understand your unique health situation.
Summary
Amyloidosis is a disease that causes abnormal amyloid protein buildup. Amyloid build-up can be dangerous and lead to organ damage and failure if left untreated. While the condition cannot be cured, symptoms cannot be treated and well-managed.
The most common types of amyloidosis are AL amyloidosis, AA amyloidosis, Abeta2m, ATTR amyloidosis, ALOC type, and wild-type. Each is unique in how it is diagnosed and treated. Further, some types have less favorable outcomes than others.
Even so, it is still possible for treatment outcomes to be positive. Your doctor can best address your concerns about treatment options or your outlook.

