
Hallmarks of Cancer 7: Genome Instability and Mutation
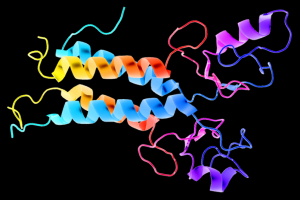

This is a 3D schematic representation (also known as a ribbon diagram) of the BRCA1 protein. BRCA1 is important for repairing damaged DNA, and BRCA1 mutations result in an increased risk for breast and ovarian cancer. Image credit: Wikimedia Commons
All cancers share ten underlying principles, also known as the Hallmarks of Cancer. You can read about the first six here. The seventh is defined as genome instability and mutation.
Cancer Cells Evolve
Not all cancer cells are equal. They vary, they compete, and the fittest survive to pass on their genes to daughter cells, which continue to vary, compete and survive. If this sounds familiar, it’s because it is; cancer cells evolve. Within the complex ecosystem that is our body, cancer cells mutate and face selective pressures as they change and adapt to their environment. The evolving cancer cell has to out-compete the normal cells surrounding it, evade attack by immune cells, escape the apoptotic machinery which causes cells to self-destruct, corrupt and co-opt otherwise loyal surrounding cells and migrate to distant parts of the body.
Sometimes, when sequences of nucleotides called genes mutate, those mutations are not corrected. It doesn’t happen often (each nucleotide has only a one-in-a-hundred-billion to a one-in-a-billion chance of being miscopied), but each time a cell divides and passes a copy of its DNA to its daughter cell, the chance of error increases. Over the average human lifetime, over one hundred million billion cell divisions occur. The average gene is thus home to somewhere between one hundred million and ten billion mutations. If just one of those mutations increases the evolutionary fitness of its cell, then that mutation will expand into many cells, increasing the probability that subsequent mutations will build in the first.
Mutations are inevitable. While mutations are still rare events, the sheer number of opportunities (i.e. cell divisions) for mutations during an average human lifetime makes the probability of a mutation for an average gene high. Image credit: Buddhini Samarasinghe
We know that cancer occurs because we can see the stepwise accumulation of mutations. If a cancer cell acquires a mutation that enables it to grow faster, or survive longer and produce more offspring than the surrounding cells without that mutation, then that cancer cell has a selective advantage. Its descendants will be fitter; they will outgrow and dominate the local tissue environment. These principles of population genetics and evolutionary biology describe the process of tumor formation and are key to understanding how cancer cells can become resistant to drug treatment; they evolve that resistance.
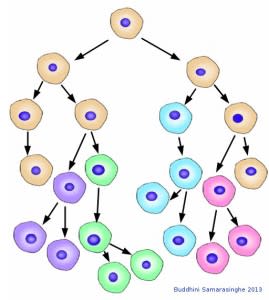
Cancer cells evolve. With each cell division, cancer cells accumulate mutations that cause variations. These variations allow cancer cells to change and adapt to survive within the complex ecosystem that is our body. In this schematic, the original cancer cell type shown in beige is no more; its descendants have all mutated and diverged, adapting and surviving with each cell division. Image credit: Buddhini Samarasinghe
Mutations 101
Mutations enable cancer cells to embark on their frenzied growth within our bodies. But what are mutations, and how do they happen? In genetics, a mutation is a change in an organism’s DNA sequence. The nucleotide letters A, T, C and G that make up our DNA can be deleted or substituted, and single or double stranded breaks can occur in the DNA molecule. Complete sections of our DNA can also be deleted or swapped with other sections. These changes can occur spontaneously or from exposure to inducers such as harmful chemicals or radiation. Our metabolic activities cause mutations all the time; oxygen, the vital molecule that helps us live, also creates dangerous DNA-damaging free radicals when metabolized by our cells. A sunny day at the beach can introduce thousands of mutations into our DNA. In fact, mutations are inevitable; each time our cells divide, the imperfect DNA replication process introduces temporary errors into our DNA. It has been estimated that all these processes can result in thousands of individual molecular lesions per cell per day. Our genome surveillance system and DNA repair mechanisms must be doing a fantastic job: based on these mutation rates, cancer should occur from the moment we are conceived.
What is our genome surveillance system? If a cell is to survive, all the different mutations must be repaired. Typically, this repair involves cutting out and re-synthesizing the damaged portion of the DNA. Although there are many different types of DNA repair, all are so important that the proteins involved can be found in everything from the humblest bacterium to our own cells.
DNA Repair Mechanisms
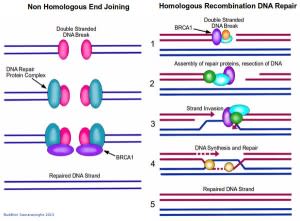
The beauty of DNA repair comes from the structure of DNA: The double helix carries two separate copies of all the genetic information in each of its two strands. When one strand is damaged, the other is used as a template to restore the sequence to the damaged strand. When both strands of the double helix are broken, however, leaving no template strand for repairs, cells may use one of two distinct alternative mechanisms to address breaks. The first, known as Non-Homologous End Joining (NHEJ), is a quick and dirty method for fixing the break; simply put, the two broken ends are brought together and joined, usually with the loss of one or two nucleotide letters at the site of joining. Although this loss results in a permanent change in the DNA sequence, it is less harmful for the cell than a persisting double-stranded break. And, since very little of our genome actually codes for protein, the resulting mutation is statistically unlikely to be of much consequence. In contrast to the error-prone NHEJ mechanism, Homologous Recombination DNA Repair exploits the fact that each cell contains two copies of each double helix (i.e. each cell has two copies of each chromosome). In this case, the repair mechanism is able to transfer nucleotide sequence information from the intact double helix to the broken one. Recombination proteins recognize regions of similarity between the two double helices and bring these regions together. Then, DNA replication proteins use the intact helix as a template to repair the broken one.
Double stranded DNA breaks are repaired by two main mechanisms: non homologous end joining (NHEJ, left), and homologous recombination DNA repair (right). BRCA1 (purple) plays an important role in both these mechanisms. In NHEJ, a double stranded DNA break is detected by DNA repair proteins which bring together the two ends and join them. This method usually results in the loss of several nucleotides, and is error prone as a result. Homologous Recombination DNA repair is more stringent. Since each cell has two copies of each chromosome, this mechanism uses one copy as a template to repair a double stranded break in the other. 1. Repair proteins assemble at the site of DNA damage. The damaged strands are indicated in red while the intact DNA strands are indicated in blue. 2. The repair proteins resect the DNA to create some room to work in. 3. The intact copy of DNA (blue strands) then 'invades' the damaged strand. 4. Repair proteins are able to use this intact copy of DNA as a template to repair the break in the damaged strand, by synthesizing DNA across the gap. 5. The double stranded DNA break is repaired, with a high level of fidelity. Image credit: Buddhini Samarasinghe
Molecular Caretakers: BRCA1 and BRCA2
Two of the most famous proteins in cancer, BRCA1 and BRCA2, play a central role in DNA repair. These proteins move to the site of DNA damage and recruit other proteins to initiate DNA repair complexes. When either BRCA1 or BRCA2 are absent as the result of a mutation, these DNA repair complexes do not form after DNA damage. Therefore, cells that are missing BRCA1 or BRCA2 are hypersensitive to agents that damage DNA, such as UV light and various chemical carcinogens. Women with an abnormal BRCA1 or BRCA2 gene, for example, have an extremely high risk of developing breast cancer and a high risk of developing ovarian cancer. As a result, many women who have a family history of breast and ovarian cancers undergo genetic testing. If they test positive for either gene mutation, they may opt to undergo a preventative mastectomy. Actress Angelina Jolie, who tested positive for a BRCA1 mutation and opted to undergo a preventative double mastectomy, recently brought this issue to the spotlight. While the publicity she generated was controversial, Jolie reduced her risk for developing breast cancer from 87 to less than five percent.
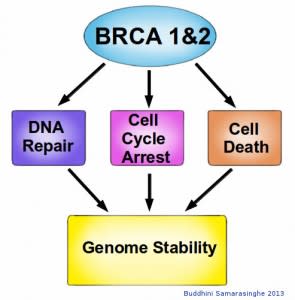
In addition to being crucial for DNA repair, BRCA1 and BRCA2 are also involved in cell cycle control. The Retinoblastoma protein, which detects damaged DNA, acts as the brake in cell cycle progression. Once they detect damaged DNA, BRCA1 and BRCA2 are crucial for the activation of these cell cycle checkpoints. BRCA1 also activates P53, a key protein involved in activating apoptosis in response to irreparable DNA damage. These essential roles for BRCA1 and BRCA2 proteins highlight their role in DNA repair, underscoring their function as molecular caretakers in our genome surveillance system.
BRCA1 and BRCA2 proteins have many roles within a cell. Although their primary role is in DNA repair, they are also involved in halting the cell cycle upon detection of damaged DNA, utilizing cell cycle checkpoints. BRCA proteins are also able to activate cell death if the DNA damage is too extensive for repair. Thus BRCA proteins are essential for maintaining the stability of our genome, and without them we are at increased risk for cancer. Image credit: Buddhini Samarasinghe
Stacking The Deck
When our genome surveillance system is compromised, the rate of mutations increases (similar to how anarchy breaks loose when local law enforcement is indisposed). The effect of a mutation is still random, but cancer cells can stack the deck in their favor when the genome surveillance system is compromised, ensuring that future mutations occur more frequently. When cancer cells evolve, there is selection for mutations that can overcome the ten anticancer defense mechanisms represented by the Hallmarks of Cancer. Mutations in Ras allow cancer cells to remain independent from growth factors. Mutations in Retinoblastoma remove the brakes, allowing for uncontrolled cell division. P53 mutations allow cells to escape cell death. All of these mutations are produced initially by direct DNA damage, and secondarily as a result of mutations in genes that increase the probability of more mutations (i.e. mutations in DNA repair). Thus, as cancer cells continue to evolve and cells with specific mutations are selected, cells that carry mutations in genes that normally function in maintaining the stability of the genome are also selected. Genomes vary widely between different tumor types, but nearly all of them have DNA repair defects. Thus, genome instability and mutation are essential to tumor progression.
Follow Scientific American on Twitter @SciAm and @SciamBlogs. Visit ScientificAmerican.com for the latest in science, health and technology news.
© 2013 ScientificAmerican.com. All rights reserved.
